Amyloidose des Herzens – neue Therapiemöglichkeiten
Amyloidose ist eine seltene Erkrankung, bei der Eiweiße im Körper ihre regelrechte Struktur und damit auch ihre ursprüngliche Funktion verlieren. Die abnormen Eiweißmoleküle können sich in weiterer Folge zu abnormen Fasern (sog. Fibrillen) zusammenlagern und sich in verschiedenen Geweben im Körper ansammeln. Diese Ansammlungen zerstören die Architektur des betroffenen Gewebes und beeinträchtigen die Funktion des Organs. Die Symptome der Amyloidose hängen davon ab, welche Organe betroffen sind. Zu den häufig involvierten Organen gehören das Herz, die Nieren und das Nervensystem. Die Ursache für das Vorhandensein einer Amyloidose ist in den meisten Fällen unbekannt, es gibt jedoch Varianten der Erkrankung, die vererbt werden können.
Die Ursache einer Amyloidose ist meist unbekannt, es gibt jedoch Varianten, die vererbt werden können.
Bei den Amyloidosen mit Beteiligung des Herzens wird heutzutage mit Abstand am häufigsten Transthyretin (TTR) als ursächliches Eiweiß nachgewiesen. TTR ist ein Eiweiß, das hauptsächlich in der Leber gebildet wird und eine entscheidende Rolle beim Transport von Vitamin A und Schilddrüsenhormonen spielt. TTR besteht aus vier Untereinheiten, die normalerweise miteinander verbunden sind. Im Rahmen der Transthyretin-Amyloidose (sog. ATTR-Amyloidose) können sich diese Untereinheiten jedoch voneinander trennen und abnorme Fibrillen bilden, die sich im Körper ablagern. Die kardiale ATTR-Amyloidose tritt fast immer sporadisch bei Männern ab dem 60. Lebensjahr auf (wtATTR). Die erbliche Form (vATTR) ist in Österreich äußerst selten.
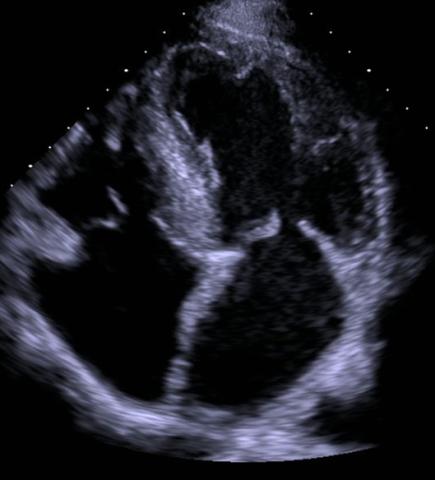
Typisches Echokardiographisches Bild bei Amyloidose
Die ATTR-Amyloidose beschränkt sich nicht auf das Herz, sondern kann auch das Nervensystem und manche Weichteilgewebe betreffen. Deshalb treten neben den typischen Beschwerden der Herzschwäche häufig weitere Auffälligkeiten auf, die als Warnsignale für eine Amyloidose gelten. Hierzu gehören u.a. das Vorhandensein eines Karpaltunnelsyndroms oder eine Einengung des Spinalkanals (sog. Spinalkanalstenose). vATTR, die angeborene Form der ATTR-Amyloidose, kann auch primär das Nervensystem betreffen und mit Gefühlsstörungen sowie Muskelschwäche einhergehen.
Die Amyloidose des Herzens bewirkt eine abnorme Verdickung der Herzwand, die durch die Einlagerung von TTR-Fibrillen verursacht wird. Durch diesen Prozess verliert das Herz nach und nach seine Dehnbarkeit und kann sich nicht mehr regelrecht füllen. In fortgeschrittenen Stadien büßt es darüber hinaus auch an Pumpkraft ein. Die ATTR-Amyloidose führt unbehandelt zu einer Herzschwäche mit eingeschränkter Leistungsfähigkeit, Kurzatmigkeit bei Belastung und Wassereinlagerungen in den Beinen. Des Weiteren kann durch die Ansammlung von abnormen TTR-Eiweißen das Erregungsleitungssystem des Herzens gestört werden, wodurch das Entstehen von Herzrhythmusstörungen begünstigt wird. Dies kann sich typischerweise in Form von Überleitungsstörungen zwischen den Vorhöfen und Kammern des Herzens manifestieren (sog. AV-Block).
Seit einigen Jahren sind neben den allgemeinen Behandlungsmöglichkeiten der Herzschwäche auch spezifische medikamentöse Therapieoptionen verfügbar, die in den Entstehungsprozess der abnormen TTR-Fibrillen eingreifen. Eine dieser spezifischen Therapieoptionen ist die Behandlung mit einem TTR-Stabilisator. Dieses Medikament bindet an das TTR-Eiweiß und stabilisiert dessen Struktur, um das Auseinanderfallen der Untereinheiten zu verhindern. Dadurch wird die Bildung der abnormen TTR-Fibrillen verhindert und das Fortschreiten der Amyloidose verlangsamt. In klinischen Studien konnte belegt werden, dass die TTR-Stabilisierung das Risiko für ein Versterben an der kardialen Amyloidose bzw. für ein Voranschreiten der Herzschwäche deutlich senkt. Eine weitere vielversprechende Option in der Behandlung dieser Erkrankung sind RNA-Interferenz-Therapien, die darauf abzielen, die Produktion von TTR zu reduzieren. Diese Therapie ist aktuell nur für PatientInnen, die an der erblich bedingten Form der ATTR-Amyloidose mit einer Beeinträchtigung des Nervensystems erkrankt sind, verfügbar. Aufgrund dieser krankheitsspezifischen Behandlungsmöglichkeiten hat die Ursachenabklärung bei Herzschwäche-PatientInnen einen hohen Stellenwert, um bereits in frühen Stadien der Erkrankung nicht nur die Symptome einer Herzschwäche zu behandeln, sondern auch den zugrundeliegenden Krankheitsprozess zu modulieren.

Tipps
- Die Behandlung der Herz-Amyloidose ist umso effektiver, je früher die Krankheit diagnostiziert wird.
- Die Herz-Amyloidose geht häufig mit Karpaltunnelsyndrom, Spinalkanalstenose und Bizepssehnenruptur einher.
Die Selbsthilfegruppe „Leben mit Amyloidose - Amyloidosis Austria“ hat es sich zum Ziel gesetzt, einen wesentlichen Beitrag zur frühestmöglichen Diagnose der Erkrankung zu leisten. Für die erfolgreiche Behandlung ist die Früherkennung - wie bei allen lebensbedrohlichen Krankheiten – wichtig!
Priv.-Doz. DDr. Nicolas Verheyen
Dr. Verheyen ist Facharzt an der Klinischen Abteilung für Kardiologie der Medizinischen Universität Graz. Als verantwortlicher Facharzt der Ambulanz für Hypertrophe Kardiomyopathien beforscht er gemeinsam mit seinem Team klinisch-wissenschaftliche Aspekte von Kardiomyopathien mit hypertrophem Phänotyp. Schwerpunkte sind die Verbesserung diagnostischer und therapeutischer Algorithmen.

Dr. med. univ. David Zach
Dr. Zach ist derzeit in der klinischen Forschung an der Abteilung für Kardiologie der Medizinischen Universität Graz tätig. Sein Forschungsschwerpunkt liegt im Bereich der hypertrophen Kardiomyopathien sowie deren Phänokopien. Dieser hat sich bereits im Rahmen von Forschungsprojekten während des Studiums der Humanmedizin herauskristallisiert und wird nun im Zuge des Doktorats konsequent weiterverfolgt, wobei hier im Speziellen die zugrundeliegenden Mechanismen der LVOT-Obstruktion im Fokus stehen.