Hypertrophe Kardiomyopathie – Diagnostik und Therapieangebot
Die hypertrophe Kardiomyopathie (HCM) ist eine isolierte Erkrankung des Herzmuskels. Bedingt durch genetische Veränderungen (sog. Mutationen) sind Herzmuskelzellen bei dieser Erkrankung „fehlprogrammiert“, ohne Anlass zu wachsen. Die Mutation kann entweder von einem Elternteil geerbt worden sein, oder (in ca. 40%) spontan beim Betroffenen neu auftreten. In der Allgemeinbevölkerung ist etwa jeder Fünfhundertste von der Erkrankung betroffen, wobei diese häufig erst spät oder auch gar nicht diagnostiziert wird. Damit stellt die HCM die häufigste genetisch bedingte Herzmuskelerkrankung dar.
Eine HCM liegt vor, wenn die Wand der linken Herzkammer auf mehr als 14mm verdickt ist (normalerweise ist der Herzmuskel maximal 10mm dick), ohne dass eine andere plausible Ursache, wie beispielsweise Bluthochdruck, ein Herzklappenfehler oder eine Speichererkrankung, gefunden werden kann. Zur Abklärung einer Herzmuskelverdickung können neben dem Herzultraschall, auch eine Magnetresonanztomographie des Herzens und eine genetische Testung notwendig werden.
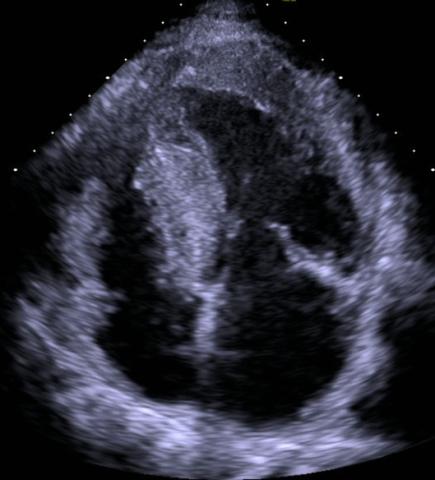
Typisches Echokardiographisches Bild bei Hypertropher Kardiomyopathie
Die klinischen Folgen der Erkrankung variieren von Person zu Person. MutationsträgerInnen können lebenslang gesund bleiben oder „nur“ eine Herzmuskelverdickung entwickeln, ohne dabei Beschwerden zu verspüren. Es kann aber auch zu einer Verschlechterung der Herzfunktion, zu Herzrhythmusstörungen oder zu einer Verengung des Ausflusstrakts der linken Herzkammer (sogenannte LVOT-Obstruktion) kommen. Diese Komplikationen der HCM lassen sich heutzutage gut diagnostizieren und effektiv behandeln. Adäquat therapierte HCM-PatientInnen haben heutzutage eine normale Lebenserwartung. Dies beruht auf mehreren Faktoren:
- Das Risiko für einen plötzlichen Herztod lässt sich mittels systematischer Erhebung der Krankengeschichte, sowie durch Langzeit-EKG, Herzultraschall und Magnetresonanztomographie gut abschätzen. Folglich können gezielt jene HCM-PatientInnen identifiziert werden, die von einem implantierbaren Defibrillator (sog. ICD-Therapie) profitieren.
- Bei Vorliegen von Vorhofflimmern kann durch Blutverdünnung (sog. Antikoagulation) das Risiko für das Erleiden eines Schlaganfalls deutlich gesenkt werden.
- Bis zu zwei Drittel der betreuten HCM-PatientInnen leiden an einer LVOT-Obstruktion. Diese führt bei körperlicher Belastung zu Kurzatmigkeit oder Schwindel, bis hin zum Bewusstseinsverlust. Bestimmte Situationen wie Flüssigkeitsverlust (z.B. Schwitzen, Durchfall), schwere Mahlzeiten oder Alkoholkonsum verstärken typischerweise diese Beschwerden. Durch bestimmte Medikamente wie Betablocker oder Calcium-Antagonisten kann häufig eine Symptomverbesserung erzielt werden. Durch invasive Eingriffe (interventionelle Alkoholseptumablation, chirurgische Myektomie) kann an erfahrenen Zentren bei einem vertretbar niedrigen Eingriffsrisiko meist eine Symptomfreiheit erreicht werden. Darüber hinaus wird gegen Ende des Jahres 2023 in der EU die Zulassung von Mavacamten erwartet. Dies ist ein neuartiger Wirkstoff, der in klinischen Studien eine exzellente Wirksamkeit bei LVOT-Obstruktion gezeigt hat.
- Im Verlauf der Erkrankung kann die Herzleistung abnehmen und sich eine Herzschwäche entwickeln. Auch hierfür stehen inzwischen sehr gut wirksame Medikamente zur Verfügung. Bei Versagen der medikamentösen Therapie wird nur in sehr seltenen Fällen die Herztransplantation als letzte therapeutische Option benötigt.

Tipps
- Kurzatmigkeit und Schwindel bei körperlicher Belastung sind bei PatientInnen mit hypertropher Kardiomyopathie häufig Ausdruck einer Ausflusstraktobstruktion (LVOT-Obstruktion).
- PatientInnen mit hypertropher Kardiomyopathie sollten an einem Zentrum behandelt werden, damit Komplikationen rechtzeitig erkannt und behandelt werden können.
Priv.-Doz. DDr. Nicolas Verheyen
Dr. Verheyen ist Facharzt an der Klinischen Abteilung für Kardiologie der Medizinischen Universität Graz. Als verantwortlicher Facharzt der Ambulanz für Hypertrophe Kardiomyopathien beforscht er gemeinsam mit seinem Team klinisch-wissenschaftliche Aspekte von Kardiomyopathien mit hypertrophem Phänotyp. Schwerpunkte sind die Verbesserung diagnostischer und therapeutischer Algorithmen.

Dr. med. univ. David Zach
Dr. Zach ist derzeit in der klinischen Forschung an der Abteilung für Kardiologie der Medizinischen Universität Graz tätig. Sein Forschungsschwerpunkt liegt im Bereich der hypertrophen Kardiomyopathien sowie deren Phänokopien. Dieser hat sich bereits im Rahmen von Forschungsprojekten während des Studiums der Humanmedizin herauskristallisiert und wird nun im Zuge des Doktorats konsequent weiterverfolgt, wobei hier im Speziellen die zugrundeliegenden Mechanismen der LVOT-Obstruktion im Fokus stehen.