Lungenhochdruck (pulmonale Hypertension)
Lungenhochdruck (pulmonale Hypertension, oder pulmonale Hypertonie, PH) bezeichnet eine Drucksteigerung und Widerstandserhöhung im kleinen Kreislauf. Der kleine Kreislauf oder Lungenkreislauf besteht aus der rechten Pumpkammer des Herzens, den Lungenarterien, den Lungenkapillaren und den Lungenvenen, die sich in den linken Herzvorhof entleeren (Abbildung 1 und 2). Lungenhochdruck entsteht durch eine Erkrankung der Lungengefäße (sogenannter prä-kapillärer Lungenhochdruck, Abbildung 1), oder durch einen Überdruck im linken Herzen, die über den linken Vorhof in die Lungenvenen und Lungenarterien zurück fortgeleitet wird (post-kapillärer Lungenhochdruck, Abbildung 2).
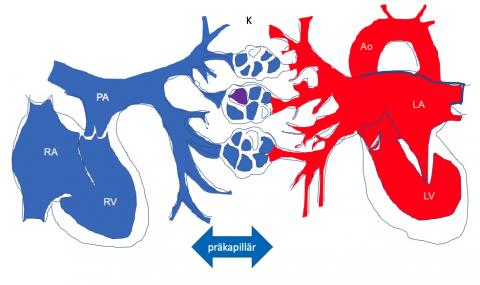
Abbildung 1 Prä-kapillärer Lungenhochdruck
Drucksteigerung und Widerstandserhöhung im kleinen Kreislauf entstehen durch eine Erkrankung der Lungengefäße kurz vor und im Kapillargebiet, sowie kurz nach dem Kapillargebiet (von den Pfeilspitzen begrenzt).

Abbildung 2 Post-kapillärer Lungenhochdruck
Drucksteigerung und Widerstandserhöhung im kleinen Kreislauf entstehen durch eine Fortleitung erhöhten Drucks vom linken Vorhof (Pfeil). Die weißen Pfeile bezeichnen die Vergrößerung und Volumenzunahme des linken Vorhofs, die typisch ist für post-kapillären Lungenhochdruck.
K=Kapillargebiet, RA=rechter Vorhof, RV=rechte Pumpkammer, PA=Pulmonalarterie, PV=Lungenvene, LA=linker Vorhof, LV=linke Pumpkammer, Ao=Aorta, große Körperschlagader
Es ist sehr wichtig, diese beiden Formen von Lungenhochdruck zu unterscheiden, weil die prä-kapilläre Form mit Medikamenten für Lungenhochdruck behandelbar ist, aber nicht die post-kapilläre Form. Die Unterscheidung ist nur mit einer Rechtsherzkatheteruntersuchung möglich. Prä-kapillärer Lungenhochdruck ist eine seltene Erkrankung (Orphan Disease), weil sie nur bei 5 Menschen pro 1 Million pro Jahr neu auftritt. Prä-kapillärer Lungenhochdruck hat unbehandelt eine schlechte Prognose mit einer mittleren Überlebenszeit von nur zweieinhalb Jahren. Post-kapillärer Lungenhochdruck ist dagegen eine häufige Erkrankung, die besonders bei Linksherzerkrankungen auftritt. Weltweit und auch in Österreich sind Patientenselbsthilfegruppen etabliert um die Diagnosen und Behandlungen, die bei Lungenhochdruck meist sehr umfangreich sind, zu unterstützen. Die Medizinische Universität Wien, Graz und Innsbruck, und das Krankenhaus der Elisabethinen Linz verfügen über ausgewiesene Expertenzentren für Lungenhochdruck. Das Wiener Zentrum ist auch mit einem Lungentransplantationszentrum und einem Zentrum für die Behandlung der chronisch thromboembolischen pulmonalen Hypertension (CTEPH) ausgestattet.
Lungenhochdruck ist heute kein Todesurteil mehr
Die chronisch thromboembolische pulmonale Hypertension (CTEPH) ist eine seltene Lungengefäßerkrankung, die durch eine Verengung der großen Lungenarterien mit alten Gerinnseln (meist nach Lungenembolien) charakterisiert ist. Die Erkrankung ist heilbar und die chirurgische pulmonale Endarterektomie stellt hierbei die Therapie der ersten Wahl dar. Bei dieser Operation wird das verhärtete alte Gerinnsel durch den Chirurgen aus den Gefäßen herausgenommen. Leider kommen aber bis zu 50% der Patienten nicht für das operative Verfahren in Frage, und bei etwa einem Drittel der Patienten ist durch die Operation auch keine ausreichende Senkung des Lungengefäßdrucks zu erzielen. Für diese Patienten stehen seit einigen Jahren neben der Gerinnungshemmung (z.B. Marcoumar) medikamentöse gefäßerweiternde Behandlungen zur Verfügung (Adempas und Trepulmix, das unter die Haut appliziert wird) und zusätzlich auch die interventionelle Behandlung mittels Ballondehnung. Die Ballondehnung ist wirksamer als Medikamente alleine, aber oft ist eine Kombination von Ballondehnung mit Medikamenten notwendig. Für CTEPH gibt es also ausgezeichnete Therapien, die heutzutage zur Normalisierung der Lebenserwartung führen können.

Für die Behandlung der pulmonal-arteriellen Lungenhochdruckerkrankung (pulmonal-arterielle Hypertonie, PAH), die den Prototyp der prä-kapillären Lungenhochdruckerkrankung darstellt, stehen medikamentöse gefäßerweiternde Behandlungen zur Verfügung. Bei der PAH kommt es durch fortschreitende Verengung der Lungengefäße zu einer Verkleinerung des Kapillargebiets der Lunge. In Österreich behandeln wir PAH mit unterschiedlichen gefäßerweiterenden Medikamenten (Endothelin-Rezeptor-Blocker Macitentan (Opsumit) und Ambristentan (Volibris), Phosphodiesterasehemmer Sildenafil und Tadalafil, Hemmer der löslichen Guanylatzyklase Riociguat (Adempas) und Prostazyklin-Analoga (Trisuva, Flolan oder Dynovas oder Veletri, Ilomedin oder Ventavis, Selexipag als Uptravi). Diese Medikamente sollen als Ersttherapien kombiniert angewendet werden. Das Wiener Zentrum glaubt an die HIT-HARD-AND-EARLY Behandlung, das heißt, dass man frühzeitig Prostazykline gibt. Die Pumpentherapien mit Prostazyklinen sind aber herausfordernd und verlangen eine spezielle Betreuung. Die Lungentransplantation steht für Patient:innen im fortgeschrittenen Erkrankungsstadium zur Verfügung.

Patienten-Tipps
- Lungenhochdruck ist heute kein Todesurteil mehr
- In Österreich werden bei Verdacht auf Lungenhochdruck Patient:innen an Expertenzentren überwiesen (Lungenhochdruckambulanzen)
- In Österreich kann Lungenhochdruck mit allen zugelassenen Medikamenten behandelt werden.
- Betreuung von Patienten mit Lungenhochdruck in Expertenzentren ist lebenswichtig und sinnvoll.
Univ. Prof. Dr. Irene Lang
Irene M. Lang ist eine klinische Kardiologin and Professorin für Gefäßbiologie an der Medizinischen Universität Wien. Sie wurde im Jahr 2005 von der World Medical Association als “Caring Physician of the World” nominiert. Sie leitet eine Ambulanz für Lungengefäßerkrankungen und ist aktive interventionelle Kardiologin. Sie leitet ein Forschungsprogramm über die Biologie von Gefäßverschlüssen mit besonderem Schwerpunkt auf Lungengefässerkankungen. Prof. Lang ist Mitglied der Österreichischen und Europäischen Gesellschaft für Kardiologie und des Senats der Medizinischen Universität Wien. Sie ist Mit-Herausgeberin von internationalen Fachzeitschriften wie Pulmonary Circulation, European Heart Journal und Atherosclerosis.
Stellvertretend für das Lungenhochdruck Team im AKH Wien: Univ Doz Dr Nika Skoro-Sajer, Dr Roela Sadushi-Kolici, Univ Doz Dr Christian Gerges, Dr Ioana Campean, Univ Doz Dr Sharokh Taghavi, Univ Doz Dr Bernhard Moser, Dr Stephan Schwarz, Univ Doz Dr Helmut Prosch, Univ Doz Dr Konrad Hötzenecker, Univ Doz Dr Peter Jaksch, Dr Lukasz Antoniewicz.